T. M. Murali's Research Interests
The functioning of a living cell is governed by intricate networks of physical, functional, and regulatory interactions among different types of molecules. Recent experimental advances have yielded unprecedented insights into the structure of these interaction networks and into patterns of molecular activity (mRNA, proteins, and metabolites) in response to different conditions. The ultimate goal of my research is to build phenomenological and predictive models of these networks by developing approaches that investigate the relationships among the molecules in a cell, how these elements are organised into functional modules, how these modules interact with each other, and how different modules become activated or de-activated in various cell states. My research group develops algorithms and computational tools based on graph theory, data mining, and machine learning to obtain system-level insights into these basic biological questions by studying them in a comparative manner, for example, across organisms, diseases, external perturbations, or cell states. This work is driven by collaborations with life science researchers spanning diverse fields including biochemistry, biophysics, infectious diseases, plant pathology, and tissue engineering. This page describes my group's main research areas and also highlights some current and former research projects.Main Research Areas
- Novel algorithms for network biology
- Discovery and analysis of molecular hypergraphs
- Synthesis of top-down and bottom-up approaches
- Systems biology of bioengineered livers
- Host-pathogen protein interactions
- Whole-genome gene function prediction
Novel algorithms for network biology
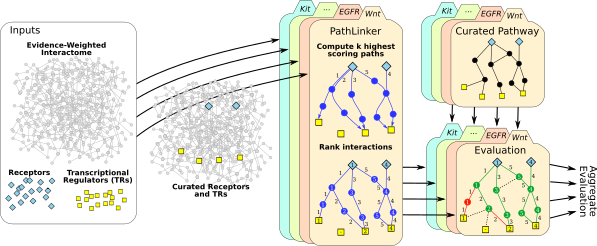
Molecular interaction networks are used pervasively in systems biology. Since these networks are complex, their analysis requires the development of novel algorithms. Our group have made several contributions in this direction. We developed one of the earliest approaches to predict gene function based on functional linkage networks. We have applied these ideas in HIV and AIDS-related research. We showed that these approaches can successfully predict human proteins that are HIV dependency factors, i.e., human proteins that HIV needs in order to replicate. We developed XTalk, the first network-based approach to predict when two signaling pathways may crosstalk. Recently, my group has developed a significant new algorithm called PathLinker to accurately reconstruct human signaling pathways. We experimentally validated a new pathway that amplifies signaling through β-catenin in Wnt signaling.Discovery and analysis of molecular hypergraphs
Molecular interaction networks are ubiquitous in computational biology. Existing methods predominantly model such networks as graphs, where each edge represents a pairwise relationship between two nodes. However, many signaling mechanisms such as complex assembly and disassembly and regulation cannot be described using these representations. My group has highlighted the underutilized role of extensions of directed hypergraphs in this context and discussed challenges and opportunities for the computational biology community. Even the notion of a path becomes non-trivial in directed hypergraphs. We have begun developing the foundational algorithms that can be meaningfully applied to signaling pathways.In related research, we have developed the novel use of hypergraphs to represent experimental uncertainty or biological variation among a group of molecules across an ensemble of graphs. We have formulated definitions of hyperedges that capture this variation in a graph ensemble and developed algorithms to compute such hyperedges. A paper from my group on this topic received the best paper award at the 2012 ACM Conference on Bioinformatics, Computational Biology and Biomedicine. The hyperedges we have discovered directly suggest groups of genes for which further experiments may be required in order to precisely discover interaction patterns. We have extended this approach to compute unstable communities in network ensembles.
Synthesis of top-down and bottom-up approaches in systems biology
Top-down approaches automatically analyze large-scale datasets for correlations between genes and proteins. Bottom-up approaches painstakingly craft detailed models that can be simulated computationally. Developing the models is a manual process that can take many years. These approaches have largely been developed separately. I am working with Dr. John Tyson (Dept. of Biological Sciences, Virginia Tech) and Dr. Jean Peccoud (Dept. of Chemical and Biological Engineering, Colorado State University) to meld the strengths of these two approaches into a single framework, thereby allowing efficient and automated data-driven analysis to augment models that can be simulated. We are using these developments to study the process of cell division in healthy cells. My group developed the Linker algorithm (a precursor to PathLinker) to analyze regulatory and physical interactomes to automatically suggest how a query protein may control a dynamic model of the budding yeast cell cycle. These suggestions have informed the development of a powerful model that can explain over 250 cell cycle mutants published in the literature and experimental validations of this model.
Systems biology of bioengineered livers
I collaborate with Dr. Padma Rajagopalan (Dept. of Chemical Engineering, Virginia Tech) on systems biology approaches to study in vitro models of the liver that she has developed. These models incorporate multiple cell types. Communications between different cell types contribute to the enhanced biological activity of these liver models. However, the precise pathways of communication remain cryptic. We are developing a combined experimental and computational strategy to discover these unknown modes of communication. In particular, I am developing novel computational techniques that analyze gene expression datasets in the context of molecular interaction networks in order to prioritize signaling pathways that would be useful to study experimentally at the protein level. This research falls under the auspices of the Center for Systems Biology of Engineered Tissues, which Dr. Rajagopalan and I co-direct. Our long term goal is to define a synthesis of systems biology and tissue engineering so that seamlessly intertwined computational and experimental models will drive the next generation of advances in both fields.Host-pathogen protein interactions
Physical interactions between pathogen and host proteins often play a critical role in initiating, sustaining, or preventing infection. A major goal of my research is the computational prediction and analysis of protein interaction networks underlying infectious diseases. I have co-developed the first technique to predict host-pathogen protein interactions. We have also developed the first global study of the landscape of human protein interacting with viruses and other pathogens, e.g., finding that viruses may have evolved to interact with human proteins that are hubs and bottlenecks in the human protein interaction network. The PIG webserver provides an interface to study these interactions. In 2010, we published one of the first large-scale datasets of human-bacterial protein-protein interactions.
Drugs for infectious diseases usually target viral and bacterial molecules. Unfortunately, viruses and bacterial pathogens become drug resistant because they mutate rapidly. Therefore, efforts to target human proteins in order to combat drug resistance are gaining currency. The recent discovery of HIV dependency factors (HDFs) is exciting for these reasons. HDFs are human proteins that when silenced block virus propagation. We have used graph-theoretic approaches to predict novel HDFs. Our results suggest that existing experimental screens for HDFs are incomplete and point to the existence of 100s of novel, undiscovered HDFs.
The precise biological roles of as many as 50% of genes in sequenced genomes is unknown or poorly known, posing a major impediment to progress in systems biology. I co-authored one of the earliest approaches to predict gene function based on functional linkage networks and later extensions
of that approach. In our approach, each node in the FLN corresponded to a gene; we labeled the node by the set of functions that annotated the gene. An edge in an FLN connected two genes if they were co-expressed and if their protein products interacted physically. We proposed an approach based on Hopfield networks to propagate functional labels in a
controlled manner across the edges of the FLN.



Gene function prediction
Current and Former Research Projects
PathLinker
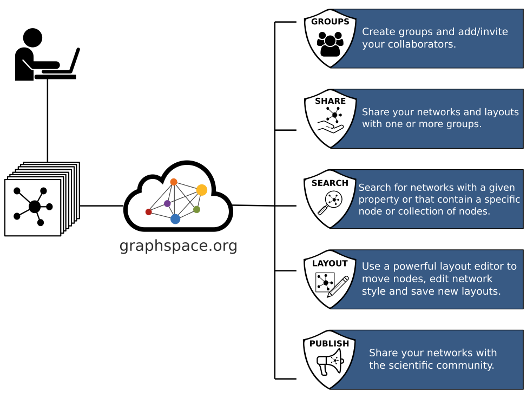
GraphCrowd
Network legos
We have developed the notion of network legos to model condition-sensitive building blocks of molecular interaction networks. A network lego has two interpretations: it is (i) a functional module of coherently-perturbed molecular interactions that is explicitly associated with (ii) a Boolean formula that denotes the set of conditions under which the module is perturbed and the set of conditions that leave the module unaffected. Our approach simultaneously generalised existing top-down methods that cluster molecular interaction networks into modules and approaches that compute the sub-network of interactions that are activated in the cell in response to a specific condition. We formulated the problem of computing network legos in terms of finding statistically-significant closed biclusters in binary matrices. In a case study of three leukemias, we used the network lego concept to automatically deduce that the Kit-receptor pathway is activated in acute myeloid leukemia (AML) but not in acute lymphoblastic leukemia (ALL) or in mixed lineage leukemia (MLL), a fact that has considerable support in the literature.

Ligand Migration Pathways in Myoglobin
Myoglobin is a small globular protein utilized by muscle cells for oxygen storage and transport. Despite nearly half a century of experimental and theoretical studies, the precise manner in which oxygen ligand moves between heme iron and solvent in myoglobin remains unresolved. We used room-temperature molecular dynamics simulations to show that there are two discrete dynamical pathways for ligand migration in myoglobin. The pathways are located in the "softer" domains of the protein matrix and go between its helices and in its loop regions. A key aspect of this work was a novel algorithm to identify cavities in static structure snapshots sterically capable of accommodating the ligand. The algorithm uses simple but highly-effective principles from computational geometry and graph theory to (i) compute cavities in each snapshot that can contain the ligand and (ii) integrate cavities across multiple snapshots to reveal dynamic migration pathways.
