THE GENERALIZED BORN APPROXIMATION
MD simulation of proteins using the new GB model
A new modification of the GB model can be used to explore protein dynamics. As an example, we have performed 8 ns. simulations of a 108-residue protein thioredoxin, protein-A (46 residues), and ubiquitin using this the new GB model. Although the simulation models the protein solvated in an infinite volume of water, the computational time is only 6 times larger than in the corresponding in vacuo simulation.
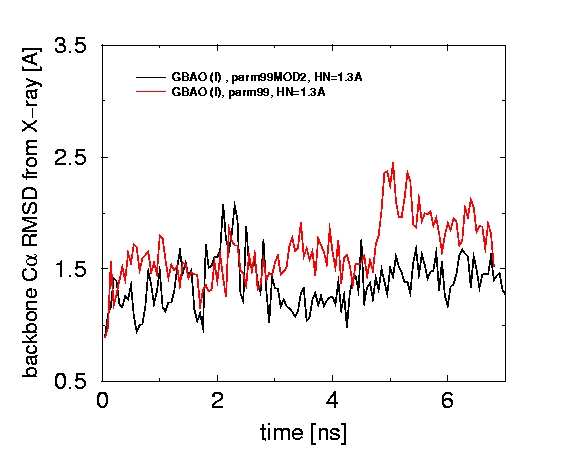
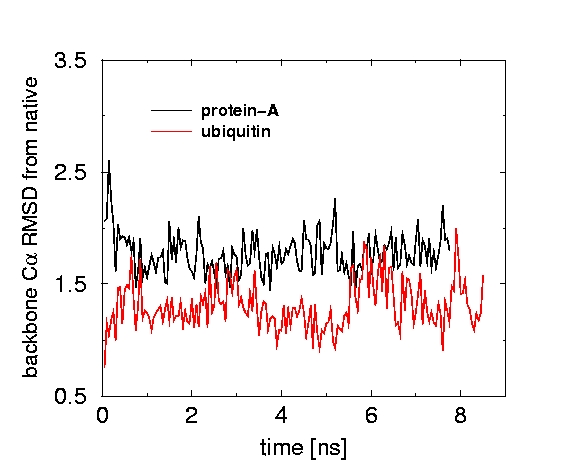
The back-bone RMSD from X-ray structures remained reasonably small in all cases (in a separate simulation that used explicit water and PME, the RMSD was 1.2 at t=6 ns).
Modeling large scale conformational changes with the new GB model
The usefulness of the GB model in simulating global conformational changes can be explored by simulating the formation of Barnase/Barstar complex from an unbound state. The results of this kind of simulation based on the new GB model are shown below. Initially, the two proteins are separated by 4.5 Angst. relative to their position in the X-ray structure of the complex. During the next 0.2 ns of the simulation, barstar docks back into barnase, and stays docked over the rest of the simulation. The decrease in the potential energy correlates well with the formation of the complex: the structure corresponding to the lowest potential energy at t=0.409 ns has backbone RMSD from the X-ray (complex) of 1.88 \AA.
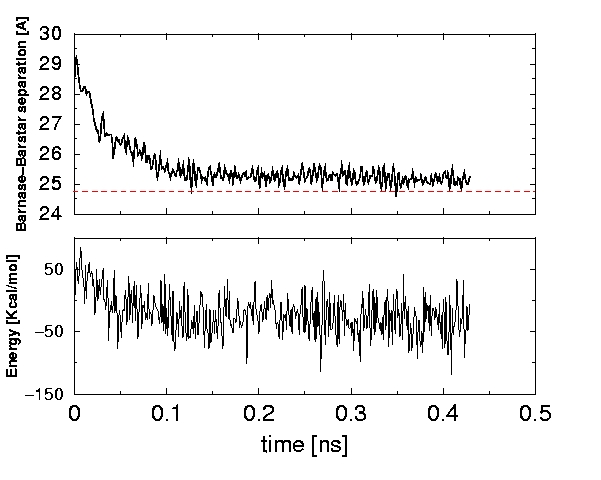
Separation between barnase and barstar, measured as the distance between their centers of mass (top), and system's potential energy (bottom) during the molecular dynamics simulation of the complex formation. GB(AO) (II) model based on modified Bondi radii set and AMBER parm99MOD2 underlying force-field is used. Red line in the upper panel indicates the distance between barnase and barstar centers of mass in the X--ray structure of the complex.
E-mail: onufriev@cs.vt.edu