PROTEIN FOLDING
ACID-UNFOLDING OF APOMYOGLOBIN
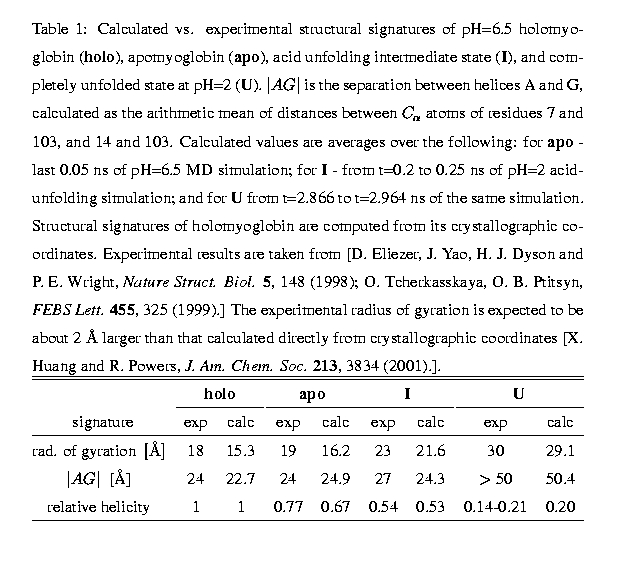
Compared to the native state, intermediate states on the protein folding/unfolding pathways are characterized by large structural fluctuations, which usually preclude resolution of the full structure of non-native states by X-ray crystallography or NMR. Under these circumstances, theoretical models may be the best alternative for elucidating their structural details. Using Molecular Dynamics simulations (MD) we model complete acid-induced unfolding of apomyoglobin -- the guinea pig in the experimental study of folding. The key point is that the simulation is at room temperature and models biologically relevant experimental conditions. No biasing potentials are used, the unfolding comes about naturally as a consequence of protonating the acidic side-chains.
Experimentally it is known that apo-myoglobin undergoes a two-phase unfolding, first to a molten globule intermediate (I-state) at about pH = 4, and finally to an unfolded state at pH = 2. In re-folding experiments, the I-state is shown to be an obligatory folding intermediate, suggesting strong similarity between acid-unfolding and re-folding pathways of apomyoglobin. We exploit this similarity to gain insight into the folding process of apomyoglobin.
>>>> Time course of the simulated acid-unfolding. >>>>
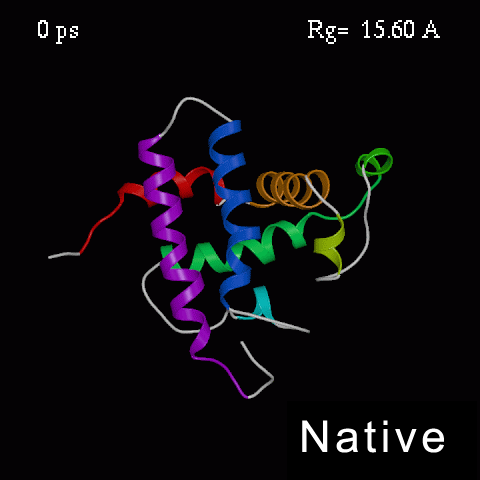
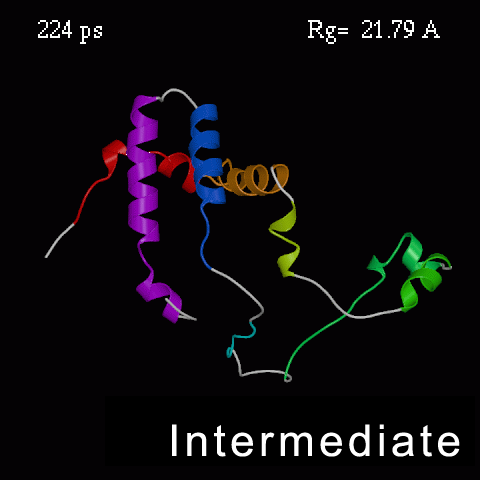
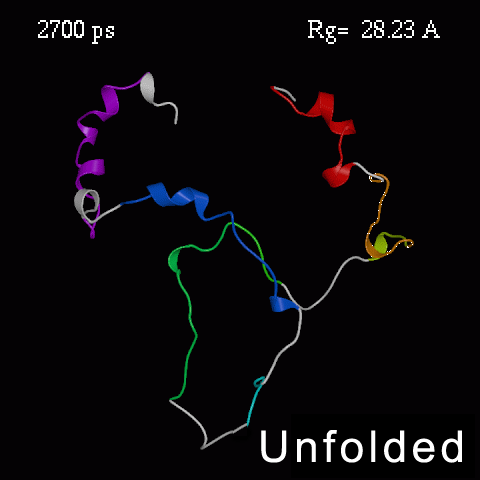
Note, that the completely acid-unfolded state retains some (~ 20%) residual secondary structure.
{kind=link}
Some of our findings:
- Structures seen on the simulated unfolding pathway are
similar to those observed in re-folding experiments, suggesting that
our results have direct implications to the
folding process.
- A near-native "dry swollen globule" state is a likely
barrier to initial
unfolding and final stages of re-folding. A number of native contacts
are disrupted,
but water does not enter the interior of the molecule,
so the
disruption of energetically favorable native
contacts is not compensated by
an increase in favorable protein-water
contacts.
- The core of the intermediate
comprising helices A, H, G, B has characteristics of
both native and molten globule states, which may resolve a long-standing
experimental controversy.
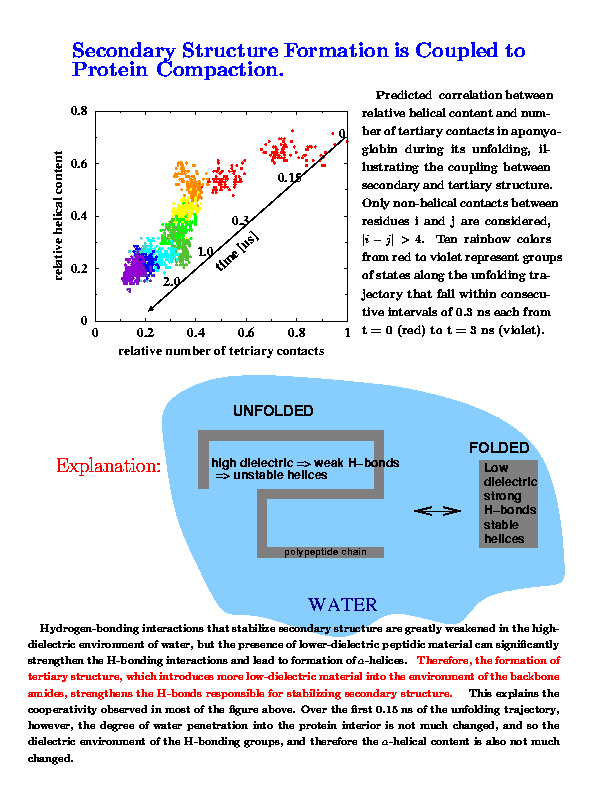
- Origin of the coupling between tertiary and secondary structure formation.
- Free energy calculations.
- Folding pathway.
Details can be found in: Alexey Onufriev, D.A. Case, and D. Bashford, "Structural details, pathways, and energetics of unfolding apomyoglobin.``, J. Mol. Biol. 325, 555-567 (2003).
E-mail: onufriev@cs.vt.edu
{kind=link}